


Vaskulitis
Eine Vaskulitis (Plural: Vaskulitiden) ist eine Entzündung der Gefäße. Vaskulitiden sind eine heterogene Krankheitsgruppe mit unterschiedlichen Ursachen sowie klinischen Ausprägungen.
Bei primären Vaskulitiden entstehen die Entzündungen und dadurch bedingte Schädigungen der Gefäßwände durch rheumatische Geschehen. Sekundäre Vaskulitiden werden durch andere Grunderkrankungen, wie Autoimmunerkrankungen oder Infektionen verursacht. Reaktionen auf bestimmte Medikamente können ebenfalls sekundäre Vaskulitiden auslösen.
Vaskulitiden können verschiedene Gefäße sowie Gefäßgrößen in beinahe jeder anatomischen Region betreffen. Abhängig von der jeweiligen Vaskulitis können Arterien, Kapillargefäße oder Venen durch entzündliche Prozesse geschädigt werden. Die Gefäßschädigungen bei Vaskulitiden können Verengungen (Stenosen), Verschlüsse, Aussackungen (Aneurysmen), aber auch eine erhöhte Durchlässigkeit zur Folge haben.
Primäre Vaskulitiden
Die Einteilung der Vaskulitiden richtet sich nach der Größe der betroffenen Gefäße, der klinischen Symptome und dem Vorliegen bestimmter Antikörper oder Immunkomplexen:
Großgefäßvaskulitiden betreffen die Aorta und Hauptäste sowie davon abgehende Arterien. Granulome in der Gefäßwand verursachen eine Narbenbildung, die dann zu einer zunehmenden Einengung des Lumens betroffener Blutgefäße führt.
- Riesenzellarteriitis: Tritt fast ausschließlich ab dem 50. Lebensjahr auf. Die Riesenzellarteriitis verursacht schmerzhafte Verdickungen der Temporalarterien. Ein Befall von Nierenarterien sowie Arm- und Beinarterien ist außerdem möglich. Die Gefahr einer Erblindung besteht, falls die A. ciliaris posterior befallen wird.
- Takayasu-Arteriitis: Betrifft vor allem Frauen unter 40 Jahren. Die Takayasu-Arteriitis ist generell selten, tritt aber in China, Indien, Japan, Thailand, Afrika und Südamerika häufiger auf.
Vaskulitiden mittelgroßer Gefäße verursachen meist Aneurysmen und Stenosen.
- Polyarteriitis nodosa (PAN): Seltene und schwerwiegende Erkrankung. PAN betrifft vor allem Männer im Alter von 40 bis 50 Jahren. Insbesondere die mittleren Arterien von Nieren, Nervensystem, Bewegungsapparat und Gastrointestinaltrakt sind entzündet und nekrotisch. Der Name „nodosa“ kommt von den knotigen Veränderungen, die aufgrund der Entzündungen entstehen.
- Kawasaki-Syndrom: Tritt im Säuglings- und Kleinkindalter auf. Betroffene Kinder leiden an fieberhaften Erkrankungen, die in lebensgefährlichen kardialen Komplikationen enden können. Mittelgroße, aber auch kleine Arterien sind nekrotisch verändert.
Die Terminologie und Einteilung von primären Vaskulitiden wurde im Jahr 2012 auf der Chapel Hill Consensus Conference (CHCC) interdisziplinär beschlossen.
Vaskulitiden der kleinen Gefäße betreffen intraparenchymale Arterien, Arteriolen, Kapillaren sowie Venen und Venolen. Die Kleingefäßvaskulitiden werden weiter unterteilt in ANCA-assoziierte Vaskulitiden und Immunkomplex-Vaskulitiden. ANCA-assoziierte Vaskuliten sind nekrotisch verändert. Im Blut sind Anti-Neutrophile cytoplasmatische Antikörper (ANCA) nachweisbar. Die vorhandenen ANCA unterscheiden sich abhängig von der vorliegenden Vaskulitis: PR-3-ANCA (cANCA), MPO-ANCA (pANCA)
- Mikroskopische Polyangiitis (MPO-ANCA): Tritt selten auf. Zu Beginn sind häufig die kleinen Kapillaren und postkapillären Venolen der Nieren und Lungen betroffen. Insgesamt hängen die Symptome von den betroffenen Organen ab.
- Granulomatose mit Polyangiitis (Wegener-Granulomatose; PR-3-ANCA): Tritt am häufigsten zwischen dem 40sten und 50sten Lebensjahr auf. Insbesondere die kleinen und mittleren Gefäße der Lunge und Niere sind nekrotisch und granulomatös verändert.
- Eosinophile Granulomatose mit Polyangiitis (Churg-Strauss-Vaskulitis; PR-3-ANCA und MPO-ANCA): Das Durchschnittsalter bei Krankheitsbeginn liegt bei 48 Jahren. Kleine und mittlere Gefäße sind granulomatös verändert mit Gewebeinfiltration durch Eosinophilie. Grundsätzlich kann jedes Organ betroffen sein, häufig beginnt die Erkrankung jedoch in der Lunge.
Immunkomplex-Vaskulitiden der kleinen Gefäße sind durch die Ablagerung von Immunkomplexen charakterisiert. Die Immunkomplexe können sowohl aus Immunglobulinen als auch Komplementkomponenten bestehen:
- Anti-GBM-Erkrankung: Die kleinen Gefäße von Lunge und Niere sind am häufigsten befallen.
- Kryoglobulinämische Vaskulitis: Kryoglobuline lagern sich vor allem in den Gefäßen der Haut, Niere und Nerven ab.
- IgA-Vaskulitis: Ist die häufigste Vaskulitis im Kindesalter. IgA-Komplexe lagern sich vor allem in der Haut, im Magen-Darm-Trakt und den Gelenken ab.
- Hypokomplementämische urtikarielle Vaskulitis: Im Blut sind Anti-C1q-Antikörper nachweisbar. Symptome betreffen meist die Gelenke, Lunge und die Augen.
Vaskulitiden variabler Gefäßgröße haben keine prädominant betroffenen Gefäßtypen. Sowohl Arterien als auch Venen können betroffen sein:
- Behcet-Erkrankung: Ist sehr selten und zeichnet sich durch rezidivierende Aphten im Mund- und Genitalbereich aus. Meist treten multiple Entzündungen auf, bei kleinen Gefäßen mit Aneurysmen.
- Cogan-Syndrom: Führt zu Vaskulitiden der Gefäße von Auge, Innenohr und Vestibulärapparat.
Einzelorganvaskulitiden können alle Organe betreffen. Der variable Befall von Arterien und Venen jeder Größe innerhalb eines Organs ist möglich:
- Kutane leukozytoklastische Vaskulitis: Ist die häufigste Form einer kutanen Vaskulitis, die potenziell auch zu Ulzerationen führen kann. Es handelt sich hierbei um eine oft in Schüben verlaufende Entzündung der kleinen kutanen Blutgefäße. Die kutane leukozytoklastische Vaskulitis tritt selten als Einzelerkrankung, sondern häufig zusammen mit ANCA-assoziierten Vaskulitiden auf. Die Ursachen für das Auftreten einer leukozytoklastischen Vaskulitis können vielfältig sein. Oft wird ein vorbestehender Infekt oder eine Reaktion auf vorhergehende Medikamenteneinnahme beschrieben. Das klinische Leitsymptom der leukozytoklastischen Vaskulitis ist die palpable Purpura. Die Purpura wird oft von Juckreiz, Schmerz oder Brennen begleitet. Im weiteren Verlauf können auch Blasen oder Nekrosen und schließlich Ulzerationen auftreten. Die Prädilektionsstellen sind die Extremitäten und insbesondere die Unterschenkel.
- Primäre ZNS-Vaskulitis: Betrifft die entzündliche Veränderung der Gefäße im zentralen Nervensystem (ZNS).
- Isolierte Aortitis: Entzündungen an der Gefäßwand der Aorta.
- Andere Einzelorganvaskulitiden: Organbezogene Entzündungen der Gefäßwände.
Außerdem können Vaskulitiden bei Systemerkrankungen, wie Lupus, Rheuma oder Sarkoidose auftreten. Vaskulitiden mit wahrscheinlicher Ätiologie entstehen als Folge einer Erkrankung oder Medikamenteneinnahme. Dazu gehören die HCV/HBV-assoziierte Vaskulitis, Syphilis-assoziierte Aortitis, Medikamenten-assoziierte ANCA-Vaskulitis und die Tumor-assoziierte Vaskulitis.
Eine durch Kokainkonsum induzierte Vaskulitis (CLIV) kann primären Vaskulitiden ähneln, weshalb ein Konsum u.U. differentialdiagnostisch in Betracht gezogen werden kann.3

Eine 25-jährige Studentin litt seit einem halben Jahr an mehreren großen Wunden am Unterschenkel und Fuß, musste wegen der Einschränkungen und starken Schmerzen ihr Studium unterbrechen. In diesem Fallbeispiel erfahren Sie, welches Krankheitsbild dahintersteckte und wie der jungen Frau schließlich geholfen werden konnte.
Jetzt Fallbeispiel lesenWeiterführende Aufzeichnungen


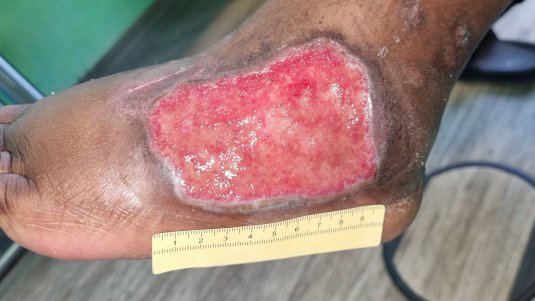
Symptome
Trotz vieler Unterschiede der Arten einer Vaskulitis gibt es doch einige typische Gemeinsamkeiten.
Allgemeine Symptome von Vaskulitiden sind Abgeschlagenheit, Fieber, Nachtschweiß und Gewichtsverlust. Bei Vaskulitiden der kleinen Gefäße entstehen häufig Purpura durch Einblutungen in die Haut. Außerdem treten oft Ulzerationen auf, die meist sehr schmerzhaft sind und rasch fortschreiten. Dadurch können auf der Wundoberfläche und an den Wundrändern häufig Nekrosen gefunden werden. Abhängig von den betroffenen Organsystemen können z.B. Kribbeln, Hörsturz, Schwindel, Sehunschärfe, Blindheit, Hämaturie oder Teerstuhl vorkommen.
Eine Vaskulitis mittlerer Gefäße hat Infarkte der betroffenen Organsysteme, wie Herz, Niere, Darm oder Extremitäten zu Folge. Bei Vaskulitiden großer Gefäße können starke Kopfschmerzen bei Befall der Karotiden und ihrer Äste auftreten. Außerdem sind das Aortenbogensyndrom, das Subclavian-Steal-Syndrom und Venenthromben mögliche Symptome.
Das Ulcus hypertonicum Martorell ist weniger bekannt als andere Unterschenkel-Ulzera und kann mit klinisch ähnlichen Erkrankungen wie einer nekrotisierenden Vaskulitis verwechselt werden.
Ulcus hypertonicum MartorellDiagnose
Die Diagnose wird auf Grundlage der Symptome, deren Schwere sowie Laborparametern gestellt.
Das diagnostische Vorgehen wird durch Verdachts- und Ausschlussdiagnosen bestimmt. Zur Labordiagnostik gehört ein Infekt- und Organscreening, die Urinanalyse und die Bestimmung von ANCAs (besonders bei Verdacht auf Kleingefäßvaskulitis. Die zu bestimmenden serologischen Entzündungsparameter BSG, CRP, Leuko- und Thrombozytose gehen mit der Akuität einer Vaskulitis oft einher und sollten wiederholt im Verlauf bei den betroffenen Patienten bestimmt werden.
Zudem sollten immer Organbiopsien entnommen werden, die sowohl konventionell als auch im Rahmen einer direkten Immunfluoreszenzdiagnostik (DIF) beurteilt werden. Der sehr einfach durchzuführende Rumpel-Leede-Test ist als Ausdruck der verminderten Kapillarresistenz und der Verlängerung der Blutungszeit bei einer Vaskulitis oft positiv. Der einfache klinische Test wird durchgeführt, indem eine Blutdruckmanschette am Oberarm angelegt und über den diastolischen Druck aufgepumpt wird. Nach fünf Minuten wird die Manschette wieder entfernt. Zeigen sich >5 Petechien/cm² Haut, so ist der Test positiv.

Allgemeine Therapieoptionen
Bei ausgeprägten Verlaufsformen kommen für die symptomatische Behandlung eine Reihe verschiedener Medikamente in Frage, die meist eine systemische Immunsuppression bewirken.
Da sekundären Vaskulitiden als Folge einer Grunderkrankung, Infektion oder Medikamenteneinnahme entstehen, müssen die zugrunde liegenden Erkrankungen therapiert bzw. verursachende Medikamente abgesetzt werden. Bei der Therapie der primären Vaskulitiden werden meist Glukokortikoide eingesetzt, um die Entzündungsreaktion im Bereich der Gefäße zu hemmen. Zusätzlich gibt es inzwischen neue Therapieansätze mit Immunmodulatoren (z.B. Blockade von TNF-alpha). Als adjuvante Behandlung wird zusätzlich symptomatisch eine Kompressionstherapie empfohlen.
Systemische Therapieoptionen sind:
- Azathioprin
- Cyclophosphamid
- Glukokortikoide
- Intravenöse Immunoglobuline
- Methotrexat
- Mycophenolat-Mofetil
- Rituximab
- TNF-α-Inhibitoren
 Schaumstoffwundauflage
Schaumstoffwundauflage
DracoFoam
 Schaumstoffwundauflage für die Ferse
Schaumstoffwundauflage für die Ferse
DracoFoam Ferse
 Polyhexanidhaltige Schaumstoffwundauflage für infizierte Wunden
Polyhexanidhaltige Schaumstoffwundauflage für infizierte Wunden
DracoFoam Infekt
 Schaumstoffwundauflage mit Haftrand
Schaumstoffwundauflage mit Haftrand
DracoFoam haft
 Dünne hydrokolloide Wundauflage
Dünne hydrokolloide Wundauflage
DracoHydro dünn
 Hydrokolloide Wundauflage
Hydrokolloide Wundauflage
DracoHydro
 Alginatkompresse
Alginatkompresse
DracoAlgin
 Polyhexanidhaltige Schaumstoffwundauflage für infizierte Wunden an der Ferse
Polyhexanidhaltige Schaumstoffwundauflage für infizierte Wunden an der Ferse
DracoFoam Infekt Ferse
 Polyhexanidhaltige Schaumstoffwundauflage mit Silikon-Haftrand für infizierte Wunden
Polyhexanidhaltige Schaumstoffwundauflage mit Silikon-Haftrand für infizierte Wunden
DracoFoam Infekt haft sensitiv
 Polyhexanidhaltige Schaumstoffwundauflage mit Haftrand für infizierte Wunden
Polyhexanidhaltige Schaumstoffwundauflage mit Haftrand für infizierte Wunden
DracoFoam Infekt haft
Literatur

Nach ihrem Studium der Biologie an der Ruhr-Universität Bochum promovierte Dr. Lorenz zum Dr. rer. nat. Seit 2012 ist sie in der medizinisch-wissenschaftlichen Abteilung bei Dr. Ausbüttel tätig, seit 2018 auch als Leiterin dieser Abteilung sowie der Forschungsabteilung.